SECOND OPINION SERIES
What Causes Undermethylation? Understanding Low SAM vs High SAH
“Undermethylation” is often treated as one condition. Biochemically, however, impaired methylation can arise through at least two major routes: the body may not produce enough SAM, its principal methyl donor, or it may accumulate too much SAH, a feedback inhibitor that slows many methyltransferase reactions.
These pathways can produce overlapping symptoms but require different clinical questions. Low SAM directs attention toward methionine, ATP, vitamin B12, folate utilization, betaine and absorption. High SAH directs attention toward homocysteine, adenosine, cellular energy, renal and hepatic clearance, creatine demand, inflammation and toxic burden.
What Causes Undermethylation? Two Primary Biochemical Pathways
Undermethylation usually develops through one of two major mechanisms: inadequate production of S-adenosylmethionine (SAM), the body’s principal methyl donor, or accumulation of S-adenosylhomocysteine (SAH), which inhibits methyltransferase activity.
The distinction matters because treatment differs. SAMe, methionine, betaine or other methylation support may help when low SAM is dominant. These same strategies may be ineffective when elevated SAH, adenosine accumulation, toxic burden, creatine demand or impaired clearance is the main bottleneck.
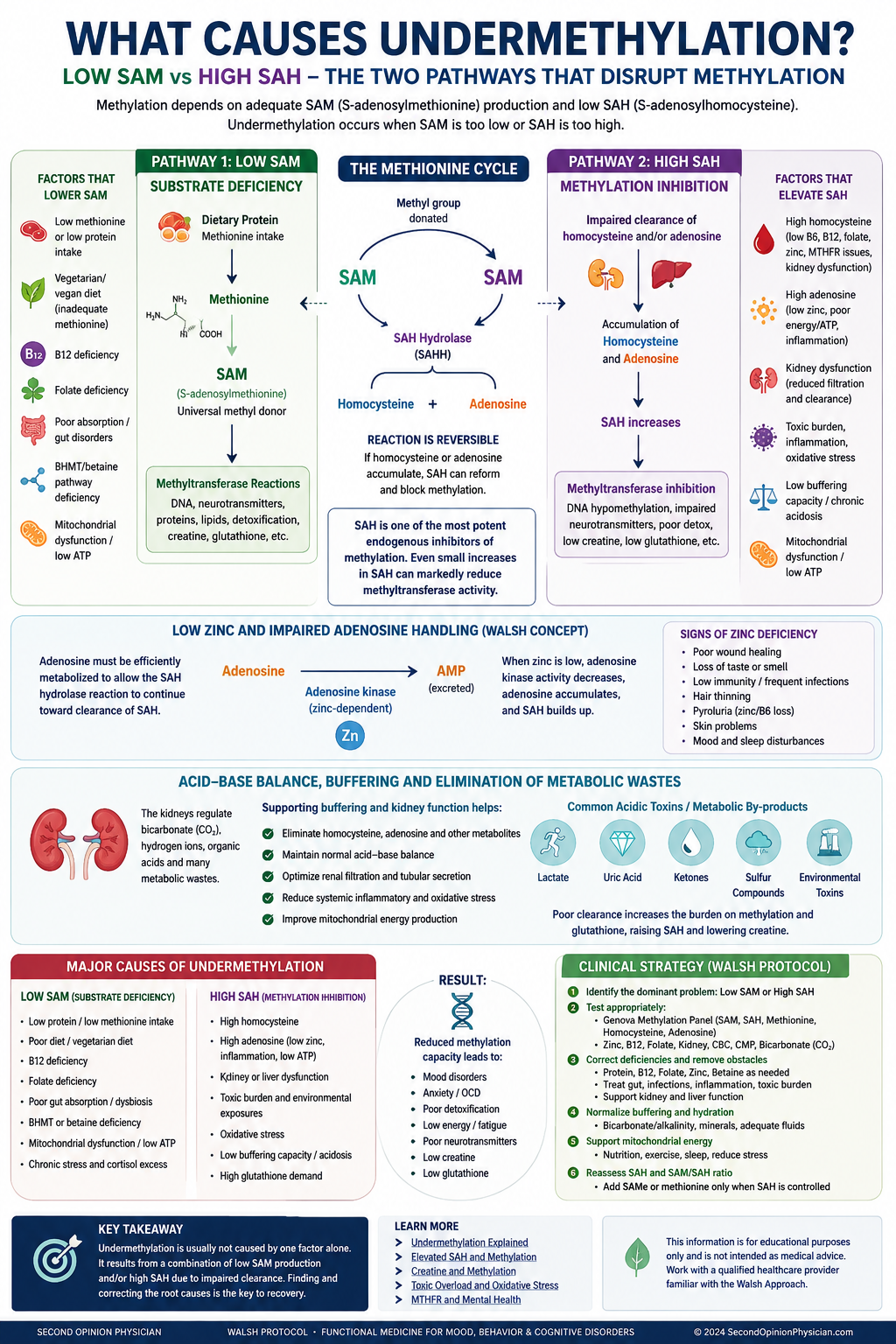
The two major biochemical tracks of undermethylation
Low SAM: not enough methyl-donor supply
SAM may be low when methionine substrate is insufficient, ATP production is impaired, remethylation pathways are limited, absorption is poor, or methyl-group demand is unusually high.
High SAH: methylation is being inhibited
SAH is produced after SAM donates a methyl group. When SAH accumulates, it can inhibit many SAM-dependent methyltransferases even if SAM itself is not severely low.
What causes low SAM?
SAM is formed when methionine combines with ATP. One-carbon metabolism then helps recycle homocysteine back to methionine through folate/B12-dependent and betaine-dependent pathways.
Low methionine or protein intake
Low protein intake, restrictive diets, malabsorption, poor digestion or inadequate essential amino-acid intake may reduce methionine availability. Vegetarian or vegan diets are not automatically deficient, but poorly planned versions can provide less methionine and creatine.
Poor absorption or gut disease
Low stomach acid, celiac disease, inflammatory bowel disease, chronic diarrhea, pancreatic insufficiency or dysbiosis may reduce absorption of protein, B12 and other cofactors needed by one-carbon metabolism.
Low ATP or mitochondrial stress
Converting methionine to SAM requires ATP. Severe cellular energy stress may therefore impair SAM production, although plasma values must be interpreted in clinical context.
Vitamin B12 or folate limitation
B12 and folate participate in remethylating homocysteine to methionine. Deficiency can raise homocysteine and impair methionine recycling.
Low betaine or BHMT pathway support
Betaine provides an alternate route for converting homocysteine to methionine, primarily in liver and kidney. Choline intake and conversion to betaine may therefore matter.
High methyl-group demand
Creatine synthesis, phosphatidylcholine synthesis, detoxification and numerous methyltransferase reactions compete for SAM-derived methyl groups.
Prenatal folate, epigenetics and undermethylation: what is known?
Dr. William Walsh has raised concern that very high methyl-donor exposure before conception or during pregnancy could influence offspring epigenetic programming in susceptible families. This remains a hypothesis rather than an established cause of Walsh-style undermethylation.
A 2020 review, Maternal Folic Acid Supplementation Mediates Offspring Health via DNA Methylation, summarizes evidence that maternal folic-acid exposure can influence offspring DNA methylation and may have both beneficial and adverse effects depending on dose, timing, baseline nutritional status and genetic susceptibility. The review supports the biological plausibility of prenatal folate altering epigenetic programming, but it does not prove that prenatal folate causes clinical undermethylation.
Research summary: Maternal Folic Acid Supplementation Mediates Offspring Health via DNA Methylation. This review describes folic acid as potentially having a “double-edged” influence on offspring health through DNA-methylation changes and calls for closer attention to dose, timing and individual susceptibility.
MTHFR and undermethylation: tested often, causal less often
MTHFR affects production of 5-methyltetrahydrofolate, which helps remethylate homocysteine through methionine synthase. Yet an MTHFR variant does not prove low SAM, high SAH or a psychiatric methylation biotype.
Functional clues can often rule in or rule out a major folate/B12 remethylation problem more quickly than a SNP report. Elevated homocysteine supports impaired remethylation or transsulfuration, while macrocytosis or an elevated MCV may suggest B12/folate deficiency—but normal homocysteine and MCV do not exclude every functional problem.
What causes elevated SAH?
After SAM donates a methyl group, it becomes S-adenosylhomocysteine (SAH). The enzyme S-adenosylhomocysteine hydrolase (SAHH/AHCY) catalyzes the reversible conversion of SAH into homocysteine and adenosine. Because the reaction is reversible, net removal of SAH depends on efficient handling of both products.
Dr. William Walsh's clinical model places special emphasis on zinc status in this process. His argument is not simply that zinc “methylates” the patient. Rather, zinc supports downstream adenosine metabolism and antioxidant defenses. If adenosine is not efficiently metabolized, it can accumulate and push the reversible SAHH reaction back toward SAH formation. In this model, low zinc can therefore contribute indirectly to persistent high SAH by impairing product clearance.
SAH must first be converted by SAHH into homocysteine and adenosine. Homocysteine must then be remethylated or directed through transsulfuration, while adenosine must be metabolized and cleared. Zinc deficiency, oxidative stress and mitochondrial dysfunction may interfere with adenosine handling. When either homocysteine or adenosine remains elevated, the equilibrium can favor regeneration of SAH, allowing SAH to continue inhibiting methyltransferases.
High homocysteine
When homocysteine is not efficiently remethylated to methionine or directed through transsulfuration, it can favor SAH accumulation.
High adenosine
Because SAH hydrolase is reversible, adenosine accumulation can also drive SAH formation. Mitochondrial energy metabolism, adenosine kinase activity and clearance may influence this balance.
Renal or hepatic dysfunction
Kidney and liver disease can disturb homocysteine, adenosine and methylation metabolites. Reduced renal filtration is a recognized contributor to elevated homocysteine.
Inflammation and oxidative stress
Chronic inflammatory and oxidative demands can redirect homocysteine toward cysteine and glutathione production and alter one-carbon metabolism.
High creatine synthesis demand
Endogenous creatine synthesis consumes a large fraction of SAM-derived methyl groups and generates SAH. Low dietary creatine or high demand may increase this burden.
Toxic burden
Mold, metals, infections, medications, gut-derived toxins and chemical exposure may increase antioxidant and detoxification demand, although the exact effect on plasma SAH varies by patient and cause.
Research: SAH as feedback inhibitor of methyltransferases · SAH hydrolase and reversible SAH metabolism
Toxic burden and undermethylation
Toxic burden can influence both tracks. It may increase the need for glutathione and other antioxidant defenses, redirect homocysteine through transsulfuration, impair mitochondrial ATP production, aggravate gut dysfunction, and increase the number of compounds requiring hepatic transformation.
This does not mean every elevated SAH result proves “toxicity.” It means toxic and inflammatory burden should be considered when methylation support fails, symptoms worsen with methyl donors, glutathione demand is high, or the patient has mold, chemical, gastrointestinal, renal or mitochondrial clues.
Homocysteine to glutathione
The transsulfuration pathway converts homocysteine toward cysteine, a limiting substrate for glutathione. Increased oxidative demand may increase pressure on this pathway.
Why sequence matters
When high SAH, inflammation or toxic burden is dominant, simply adding SAMe or methionine may not correct the bottleneck. The inhibitory or clearance problem may need attention first.
Creatine and undermethylation
Creatine synthesis is one of the body’s largest methyl-group expenses. Guanidinoacetate is methylated by SAM to form creatine, producing SAH in the process. Research estimates that endogenous creatine synthesis can consume roughly half of SAM-derived methyl groups under some conditions.
Supplemental creatine can reduce endogenous synthesis demand. This may conserve methyl groups and, in selected patients, reduce the biochemical burden on the methionine cycle. Creatine is therefore relevant to both low-SAM and high-SAH patterns, though response depends on kidney function, hydration, dose, diet and the broader clinical picture.
Research: creatine supplementation, SAM, SAH and homocysteine
Acid-Base Balance, Adenosine Clearance and Why Buffering May Support Methylation
After SAM donates a methyl group, it becomes S-adenosylhomocysteine (SAH). SAH is then metabolized through a reversible reaction involving homocysteine and adenosine. When homocysteine or adenosine accumulates, the reaction may shift back toward SAH formation. Elevated SAH can then inhibit methyltransferase activity, even when SAM is present.
Normal buffering capacity and kidney function help the body regulate bicarbonate, hydrogen ions, organic acids and metabolic waste. Chronic inflammation, infection, gastrointestinal dysfunction, diabetes, dehydration, mitochondrial stress and toxic exposure can increase the burden of frequently acidic metabolites and other compounds that must be processed and eliminated.
Supporting hydration, normal kidney filtration and adequate bicarbonate buffering may help create a more favorable environment for the removal and metabolism of adenosine, homocysteine and other metabolic by-products. This may be especially relevant when elevated SAH occurs together with a low serum bicarbonate/CO₂ level, impaired kidney function, dehydration, poor circulation, high toxic burden or evidence of mitochondrial dysfunction.
Patients with elevated SAH frequently have more than an isolated methylation problem. Toxic burden, oxidative stress and inflammation can increase the need for glutathione while also increasing demand on mitochondrial energy production, liver processing and kidney clearance. This helps explain why simply adding SAMe, methionine or methylated vitamins may fail when the underlying metabolic and elimination burden has not been addressed.
A practical evaluation may include CMP, creatinine and estimated GFR, serum bicarbonate/CO₂, electrolytes, glucose, hydration status, homocysteine, SAM, SAH, methionine and adenosine, together with clinical review of diet, kidney function, gastrointestinal health, inflammation and toxic exposure.
How do I know whether undermethylation is low SAM or high SAH?
| Finding | More suggestive of low SAM | More suggestive of high SAH |
|---|---|---|
| Direct methylation panel | Low SAM, sometimes with low methionine | Elevated SAH or reduced SAM/SAH ratio |
| Diet / absorption clues | Low protein, vegan/vegetarian imbalance, malabsorption, low B12 | May be normal, though toxic and inflammatory burden can affect appetite and absorption |
| Homocysteine | May be high with impaired remethylation or low with low substrate | Often high, but may be normal if adenosine or other factors dominate |
| Creatine demand | Can consume SAM and lower methyl-donor availability | Creatine synthesis directly generates SAH |
| Toxic / oxidative burden | May reduce ATP and increase methyl demand | May increase glutathione demand, inflammation and metabolic inhibition |
| Treatment implication | Restore substrate and cofactors cautiously | Address the SAH-driving bottleneck before simply adding more methyl donors |
Recommended undermethylation testing
Genova Methylation Panel
SAM, SAH, SAM/SAH ratio, methionine, homocysteine and related metabolites help distinguish donor shortage from product inhibition.
Walsh biotype labs
Whole-blood histamine, serum copper, ceruloplasmin, plasma zinc, urinary pyrroles and vitamin D help identify overlapping biotypes.
General metabolic testing
CBC, MCV, CMP, kidney function, bicarbonate/CO₂, B12, folate, thyroid markers and inflammatory clues can identify common contributors.
Related undermethylation and SAH articles
Undermethylation Overview
Symptoms, histamine, testing and treatment strategy.
Dr. Walsh Analysis of Elevated SAH
Why high SAH changes the interpretation of undermethylation.
Elevated SAH Therapy
Clinical strategy when SAH is the dominant methylation brake.
MTHFR and Folic Acid
Why SNP testing alone can misdirect mental-health treatment.
Creatine and Methylation
How creatine may reduce endogenous methyl-group demand.
Five Biotypes of Depression
Compare undermethylation with overmethylation, copper overload, pyroluria and toxic burden.
Dr. William Walsh: why biochemical individuality matters
This lecture provides broader context for the Walsh Approach: psychiatric diagnoses group together patients with different biochemical patterns, so the same medication or nutrient can help one patient and worsen another. The video is useful background, but the central focus of this page is the distinction between low-SAM and high-SAH methylation impairment.
Start by identifying the pathway—not guessing the supplement
SAMe, methionine, folate, methyl-B12, betaine, creatine, zinc and glutathione support can each be useful in the right context. They can also fail when the dominant bottleneck has not been identified. A practical workup combines symptoms, diet, medication history, whole-blood histamine, homocysteine, SAM, SAH, copper/zinc balance, pyrroles and toxic-burden clues.
Frequently asked questions
What is the most common cause of undermethylation?
There is no single universal cause. The most useful distinction is between inadequate SAM production and excessive SAH accumulation. Diet, absorption, B12/folate function, betaine, mitochondrial ATP, homocysteine, adenosine, creatine demand and toxic burden can all contribute.
Can toxic burden cause undermethylation?
Toxic and inflammatory burden can increase glutathione and detoxification demand, impair mitochondrial energy production and alter homocysteine metabolism. It can therefore contribute to impaired methylation, but high SAH should not automatically be labeled toxicity without broader evaluation.
Can too much prenatal folate cause undermethylation?
Folate exposure can influence developmental DNA methylation, but current human evidence does not establish recommended prenatal folic acid as a cause of Walsh-style undermethylation. The effects of excessive dose, timing and individual susceptibility remain areas for further research.
Does MTHFR cause undermethylation?
MTHFR variants can affect folate-dependent remethylation, especially when homocysteine is elevated, but they do not diagnose low SAM, high SAH or a mental-health methylation biotype.
How does creatine help undermethylation?
Supplemental creatine may reduce the need to manufacture creatine from SAM, conserving methyl groups and reducing SAH generation from endogenous creatine synthesis.
What test distinguishes low SAM from high SAH?
A plasma methylation panel measuring SAM, SAH, SAM/SAH ratio, methionine and homocysteine is the most direct way to distinguish these patterns.
Educational information only. This page does not diagnose or treat disease. Do not change prenatal vitamins, psychiatric medication or high-dose methylation supplements without qualified medical guidance.
The Walsh Protocol Explained is not an alternative belief system and not a supplement shortcut. It is a biochemical model of mental illness built from decades of laboratory data showing that people with the same psychiatric diagnosis often have fundamentally different brain chemistry.
In his lecture, William Walsh explains that modern psychiatry struggles not because clinicians lack effort, but because diagnoses such as depression, anxiety, ADHD, and schizophrenia are treated as single conditions when they are not. According to Walsh, these diagnoses group together multiple biochemical disorders, each requiring a different treatment approach.
This is the central reason nutrient therapy for depression works well for some people, fails for others, and can even make certain patients worse when applied without biochemical context.