Category: Therapeutic Diets & Ketones
Diet and nutrition form the foundation of biochemical balance, directly influencing inflammation, methylation, and brain chemistry. A Mediterranean-style diet—rich in colorful vegetables, healthy fats, fish, nuts, legumes, and modest portions of lean protein—supports stable blood sugar, provides essential minerals for enzyme activity, and lowers inflammatory load. Intermittent fasting can further improve insulin sensitivity, promote autophagy, and reduce oxidative stress, giving the body time to repair and reset detox and methylation pathways. Posts in this section highlight how diet influences neurotransmitters, copper–zinc balance, and liver function, and why processed foods, refined starches, and excess fructose can worsen symptoms of depression and fatigue. Nutrient-dense eating combined with time-restricted feeding helps optimize homocysteine metabolism, support SAM and glutathione production, and calm the inflammatory and oxidative cycles that underlie many mood and metabolic disorders. The goal is to use food and rhythm of eating as daily tools to support both mental and physical resilience.
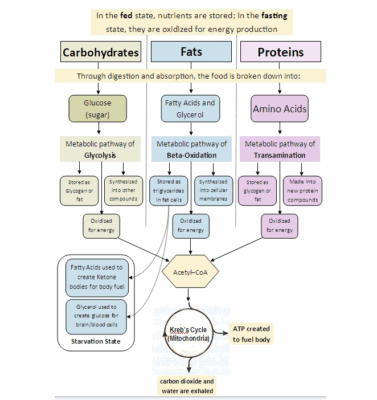
Ketogenic Therapy for OCD: Ketones not Glucose
SECOND OPINION SERIES Ketones Not Glucose: The Metabolic Mechanism Behind Ketogenic Therapy for OCD Many [...]
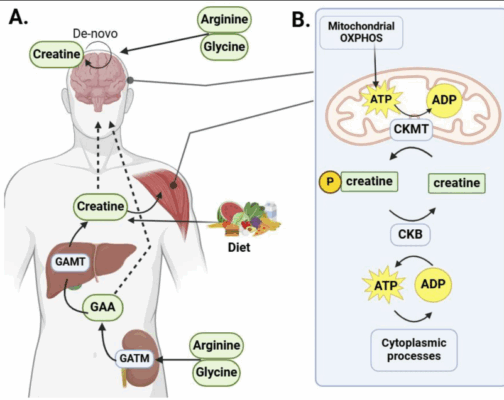
Creatine and Methylation | How Creatine Supports SAMe & Mood
SECOND OPINION SERIES Creatine Improves Methylation | Supplementation Strategy This article explains how creatine improves [...]

Raising Ceruloplasmin to Improve Copper Overload Symptoms
How to Increase Ceruloplasmin Naturally: Understanding Copper Balance, Free Copper, and Mental Health If you’re [...]

Healthy Diet for OCD & Depression
HEALTHY DIET FOR OCD AND DEPRESSION: A DIET AND RECIPE FOR MITOCHONDRIAL SUPPORT This diet [...]

OCD Methylation and Mitochondria: Ketone Ester Therapy
OCD Methylation and Mitochondria: Ketone Ester Therapy & Metabolic Treatment OCD methylation and mitochondria are [...]
2 Comments
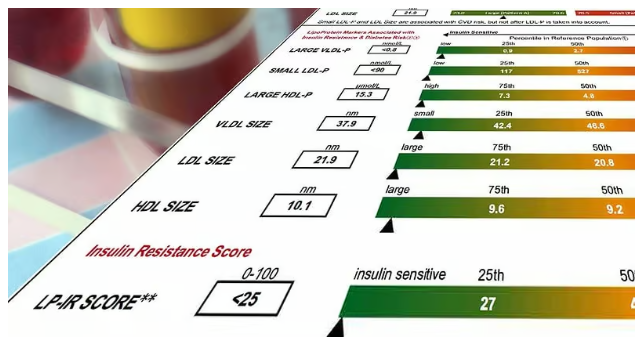
The Cholesterol Myth: What’s Good, Bad & What to Test
The Truth About Cholesterol: Why It’s Not the Villain You Think For decades, we’ve been [...]

Low Glycemic Mediterranean (Methylation) Diet
Low-Glycemic Mediterranean Diet A low-glycemic Mediterranean diet supports methylation, mood, and metabolic stability by combining [...]

Lower Cholesterol Without Statins
Lower Cholesterol Without Statins This page explains how to lower risk with diet-first changes, targeted [...]
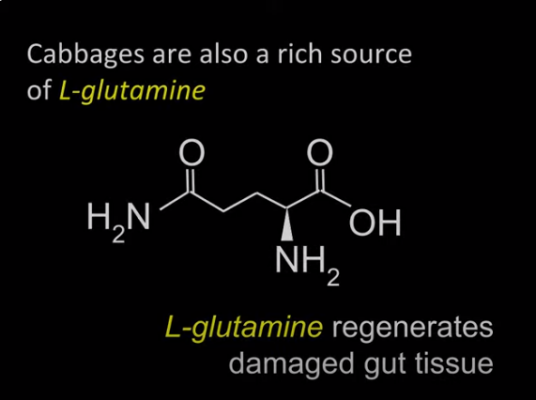
Detoxify the Liver with Vitamin U and Cabbage Juice
Vitamin U from Cabbage Juice Lower homocysteine while reducing cardiovascular inflammation and increasing production of [...]

Mastering Histamine Intolerance: Low Histamine Diet & DAO Enhancing Foods
Managing Histamine Intolerance: A Comprehensive Guide on Low Histamine and DAO Enhancing Foods Are you [...]

Migraines, Autism, Gluten & Methylation | What’s the Connection?
Migraines, Autism, and Gluten Allergies: The Underlying Role of Histamine, Undermethylation, and DAO In this [...]
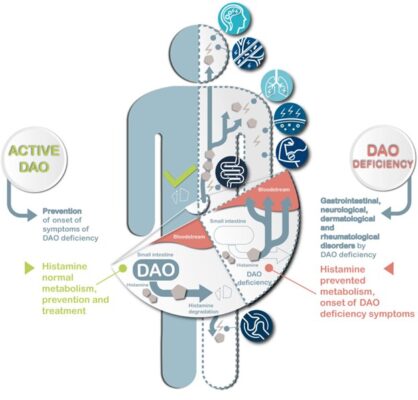
Histamine Intolerance: DAO Supplements & Testing
Histamine Intolerance: DAO Supplements & Testing Histamine intolerance can significantly impact one’s quality of life, [...]

The Link Between Obesity, Estrogen, Copper, and Anxiety
Obesity, Estrogen, and Copper: A Complex Relationship with Health Implications Obesity is a medical condition [...]
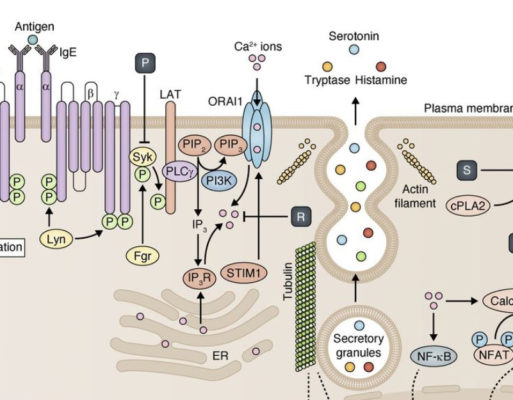
Treat Elevated Histamine, Naturally
Managing Histamine Intolerance with Methylation & Nutrient Therapy Histamine intolerance is a condition in which [...]
Foods and Supplements that Lower Plasma Histamine Levels
Foods and Antigens that Typically Raise Intestinal and Plasma Histamine Histamine rich foods: Foods that [...]
- 1
- 2