
Zinc Deficiency Meaning
Zinc deficiency refers to the condition where there is insufficient zinc in the body to carry out its numerous vital functions. Zinc is a crucial trace mineral that plays a key role in cell division, cell growth, wound healing, and the breakdown of carbohydrates. Insufficient zinc levels in the body may occur due to inadequate dietary intake, impaired absorption due to gastrointestinal disorders, excessive loss of zinc through the kidneys, or elevated physiological requirements. It's also important to highlight the role of genetic factors and the imbalance of nutrients, particularly copper overload, in contributing to zinc deficiency.
The Biochemistry of Zinc Deficiency
Zinc is an essential component of several hundred enzymes involved in the synthesis and degradation of carbohydrates, lipids, proteins, and nucleic acids, as well as in cellular metabolism. Its role in the biosynthesis of proteins is especially important, and symptoms such as white spots in nail beds can indicate poor protein synthesis resulting from zinc deficiency. Furthermore, zinc is vital for the structure and function of cell membranes, protecting cells from oxidative damage and maintaining the integrity of the immune system.
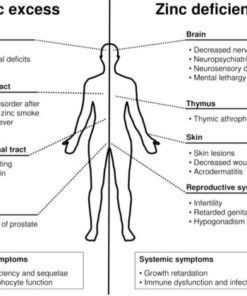
Zinc Deficiency Symptoms
Zinc deficiency can lead to a plethora of physical and psychological symptoms. These symptoms can often be subtle and easily overlooked, but they can significantly impact a person's quality of life.
- White spots in nail beds: This is a common sign of zinc deficiency. The presence of white spots can indicate poor protein synthesis due to zinc's integral role in protein metabolism.
- Poor tanning: Zinc deficiency can impair the body's ability to tan. Zinc plays an essential role in skin health, aiding in the formation of melanin, which protects the skin from the sun's harmful ultraviolet rays.
- Gastrointestinal problems: These can include a lack of appetite, nausea, diarrhea, and abdominal cramps. Zinc deficiency can lead to changes in digestive function due to zinc's essential role in the metabolism of nutrients.
- Intolerance to cheap metals: This could result from an altered immune response or a hypersensitivity reaction due to zinc deficiency.
- Premature graying: Zinc plays a vital role in maintaining hair color by playing a part in hair follicle health. A deficiency in zinc can lead to premature graying.
- Neurological issues: Anxiety, depression, and insomnia can all result from the neurotransmitter imbalances caused by a lack of zinc.
Signs of Zinc Deficiency in Females
In females, signs of zinc deficiency can often manifest in ways that can significantly affect their quality of life:
- Obesity: Zinc plays a crucial role in regulating appetite and metabolism. A deficiency in zinc can disrupt these hormones, leading to weight gain or difficulty losing weight.
- Mood disorders: Mood disorders such as depression, anxiety, and mood swings can result from the neurotransmitter imbalances caused by zinc deficiency.
Low Zinc Symptoms in Males
In males, zinc deficiency can show up as:
- Decreased libido: Zinc is integral to testosterone production, and a deficiency can impact sexual health.
- Hair loss: Zinc plays a vital role in hair health and growth. Its deficiency can lead to hair thinning or even baldness in severe cases.
Zinc Deficiency Skin Problems
Zinc is an essential mineral for skin health, and its deficiency can lead to various skin problems:
- Poor wound healing: Zinc plays a critical role in cell growth and repair, making it crucial for the process of wound healing. Deficiency in zinc can result in slow wound healing and an increased risk of infection.
- Acne: Zinc deficiency can exacerbate acne by promoting inflammation and bacterial growth in the skin.
- Dry and rough skin: Lack of zinc can affect the skin's ability to retain moisture, leading to dry and rough skin.
Foods with Zinc
Zinc is present in a variety of foods, but it's particularly abundant in protein-rich foods:
- Meat and poultry: Beef, pork, and chicken are excellent sources of zinc.
- Fish and shellfish: These are great sources of zinc, with oysters being especially rich in the mineral.
- Legumes: Lentils, chickpeas, and other legumes contain significant amounts of zinc.
- Nuts and seeds: Almonds, cashews, hemp seeds, and flax seeds are all high in zinc.
- Dairy products: Foods like milk, cheese, and yogurt provide substantial amounts of zinc.
- Whole grains: Brown rice, oats, and whole grain bread contain more zinc than their refined counterparts.
Copper Overload Due to Zinc Deficiency
Copper and zinc have a complex interplay in the body; they function as antagonists, meaning high levels of one can lower levels of the other. Zinc is necessary for the production of metallothionein, a protein that binds copper and aids in its removal from the body. When zinc levels are low, less metallothionein is produced, potentially leading to copper overload. Symptoms of copper overload can include fatigue, headaches, skin sensitivity, mood swings, and difficulty concentrating. Moreover, high levels of copper can inhibit the absorption of zinc, contributing further to zinc deficiency.
Zinc Deficiency Treatment
Treatment for zinc deficiency typically involves increasing zinc intake either through diet or supplements. It's important to remember that treatment should be under the guidance of a healthcare provider, as excessive zinc can lead to zinc toxicity, further exacerbating the delicate balance between zinc and copper. Depending on the severity of the deficiency, supplemental zinc doses may be required, along with regular monitoring of zinc levels.
Zinc Deficiency Test
The most common method of testing is through assessing plasma or serum zinc levels, which can be ordered on this website. Copper tests are also performed to assess for overload, especially in cases of suspected zinc deficiency. Here at our medical service, we provide both zinc and copper tests along with expert consultation to interpret these tests and devise an appropriate treatment plan.
The Walsh approach to treating mood disorders, which recognizes the importance of balancing nutrients like zinc and copper, further underscores the need for targeted testing and personalized treatment strategies.
It's crucial to remember that health is a lifelong commitment, and understanding your body's needs, including nutrient balances, is integral to this process. Our functional medicine telehealth services are designed to assist you in identifying and addressing any potential nutrient imbalances, promoting optimal health. Don't hesitate to reach out and prioritize your health today.
While we do not ascribe to the dosage recommended in this article, it provides useful information with respect do health conditions associated with zinc deficiency: NIH Zinc Fact Sheet for Consumers
Zinc Deficiency | Copper Overload Articles





Zinc Deficiency Testing
Other Single Item Tests

All Cognoscopy Labs
All Cognoscopy Labs
All Cognoscopy Labs
Walsh Approach Test Panels